



金针菇Flammulina filiformis (Ge, X.B. Liu & Zhu L. Yang) P.M. Wang, Y.C. Dai, E. Horak & Zhu L. Yang在中国是一种拥有悠久驯化和栽培历史的食用菌(Ge et al. 2015;戴玉成和杨祝良 2018;Wang et al. 2018),因其菌盖似铜钱,菌柄有纤毛,故又被称为毛柄金钱菌。金针菇作为一种著名的食、药两用菌,营养丰富、口感脆爽,深受消费者的青睐,其生产消费量也常年位居全球食用菌生产消费榜单的前列(Zhang et al. 2013;Kim et al. 2016)。菌柄细长,菌盖小而内卷是金针菇明显区别于其他食用菌的商品性状,是金针菇主要可食用部位(Gruen 1969;Gruen & Wu 1972)。因此,金针菇菌柄发育调控机制具有很大的研究价值。
在适当的环境条件下,内源基因和蛋白的特异性表达在金针菇菌柄生长发育过程中起到了重要作用。查美铃等(2010)通过地高辛标记的Flammutoxin基因特异性探针,发现该基因仅在金针菇菌柄部位表达。姜思源等(2018)通过实时荧光定量PCR技术鉴定得到了一个在菌柄中高表达的腺苷酸酶超家族基因Fv-Afe1。可调节细胞壁代谢活动的扩张蛋白Fv-expl1基因对于金针菇的菌柄发育也具有影响(Huang et al. 2018)。以上研究都为金针菇菌柄发育机制的深入研究提供了帮助。如果能够结合最新的组学测序技术,从整体水平上挖掘参与菌柄生长发育的特异性表达基因,将会大大促进该生物学过程的调控机理研究。
转录组学(transcriptomics),是指在整体水平上分析研究对象中基因的表达及其转录调控的学科。换言之,转录组学是在RNA水平上研究全基因组的表达情况(Velculescu et al. 1997;Thompson & Goggin 2006;Costa 2010)。转录组测序(RNA-seq)技术通过比较不同转录本之间的基因表达差异,从而分析基因表达的动态变化过程,目前已被广泛应用于生物学过程的研究中(Wang et al. 2009)。王威等(2015)通过分析金针菇质配前后的转录组,发现脂肪酸、氨基酸以及大部分糖类合成相关基因呈现上调表达,表明了金针菇异核菌丝主要进行营养物质的富集。在金针菇原基发育过程中,核糖体以及DNA复制过程中的差异基因全部呈现上调表达,而糖酵解途径中关于丙酮酸形成过程的11个差异基因呈下调表达(刘芳等 2014)。近年来,蛋白组测序技术的发展使得我们能够从蛋白水平进一步揭示重要生物过程的调控机制。运用蛋白组学技术的最新研究中,挖掘了大量参与胁迫应答、生长发育等生命活动的关键蛋白(Liu et al. 2017, 2018)。因此,转录组结合蛋白组联合分析技术能够进一步促进我们对生物重要生命活动的深入理解。
本研究基于金针菇伸长期菌柄与成熟期菌柄转录组和蛋白组测序,联合分析差异表达的基因和蛋白,以期发掘金针菇菌柄发育中起关键作用的基因及通路,从而为深入揭示食用菌菌柄发育的调控机制提供理论支撑,促进食用菌分子辅助育种技术的发展。
1 材料与方法
1.1 实验菌株
金针菇双核体菌株F19由福建省食用菌种质资源保藏与管理中心提供。
1.2 参考基因组数据
金针菇单核体菌株L11基因组数据由福建农林大学菌物研究中心提供,数据已提交至DDBJ/EMBL/GenBank(
1.3 总RNA提取及转录组测序
图1

图1
金针菇菌柄样品
A:伸长期;B:成熟期
Fig. 1
Stipes of Flammulina filiformis.
A: Elongation stage; B: Mature stage.
委托上海欧易生物医学科技有限公司将检测合格的总RNA进行建库后,使用Illumina HiSeqTM 2500测序仪进行测序,产生2×125bp的双末端数据。每个样品设置3个生物学重复。
1.4 差异表达基因分析
使用参考L11基因组序列以及注释文件作为数据库,采取序列相似性比对的方法鉴定出各蛋白编码基因在各样本中的表达丰度。使用htseq-count软件获取每个样本中比对到蛋白编码基因上的reads数,cufflinks软件来计算蛋白编码基因的表达量FPKM(fragments per kb per million reads)值(Roberts et al. 2011a, 2011b;Anders et al. 2015)。差异表达基因(differentially expressed genes,DGE)的筛选使用基于测序的检测差异基因的方法所开发的算法来筛选两样本间的差异表达基因(Audic & Claverie 1997),通过控制FDR(false discovery rate)来决定P‐value的域值,把差异表达的基因的显著度和可信度区间定义为FDR≤0.001,Foldchange ≥2倍。
将筛选到的全部差异表达基因进行GO(gene ontology)功能注释与聚类,以KEGG (Kyoto Encyclopedia of Genes and Genomes)数据库中真菌数据作为代表集对差异表达基因进行pathway富集分析(Kanehisa et al. 2008)。
1.5 总蛋白提取与蛋白组测序
使用与转录组测序相同的样品,同时委托上海欧易生物医学科技有限公司进行蛋白组测序。主要流程为:提取样品中总蛋白,取出一部分进行蛋白浓度测定及SDS-PAGE检测,另取部分进行胰蛋白酶酶解及TMT标记,然后取等量的各标记样品混合后进行色谱分离,最后对样品进行LC-MS/MS分析及生物信息学数据分析。每个样品设置3个测序重复。
1.6 差异表达蛋白分析
在可信蛋白基础上,选取两个标准计算样品间的差异。其中Foldchange用来评估某一蛋白在样品间的表达水平变化倍数;经t-test检验计算的P-value展现样品间差异的显著程度。差异筛选条件:Foldchange≥2倍,且P-value<0.05。得到差异表达蛋白之后,对差异蛋白进行GO/KEGG富集分析,对其功能进行描述。
2 结果与分析
2.1 测序数据统计
转录组测序每个样品数据过滤后得到clean_bases为2.5G。每个样品定位到参考基因组的总reads均达到88.35%以上,单一定位reads数均达到87.67%以上(表1)。测序质量较好,能够用于后期的数据分析。转录组数据已提交至NCBI(
表1 reads与参考基因组比对率统计
Table 1
| 样品 Sample | 伸长期菌柄 Elongation stipe | 成熟期菌柄 Mature stipe | ||||
|---|---|---|---|---|---|---|
| ES-1 | ES-2 | ES-3 | MS-1 | MS-2 | MS-3 | |
| 总reads数 Total reads | 16 136 676 | 16 949 982 | 18 445 804 | 17 193 950 | 15 340 044 | 15 959 110 |
| 总定位reads数 Total mapped reads | 14 262 342 (88.38%) | 15 068 103 (88.90%) | 16 297 184 (88.35%) | 15 290 110 (88.93%) | 13 670 715 (89.12%) | 14 233 650 (89.19%) |
| 多重定位 Multiple mapped | 102 028 (0.63%) | 121 202 (0.72%) | 125 696 (0.68%) | 112 767 (0.66%) | 99 820 (0.65%) | 110 493 (0.69%) |
| 单一定位 Uniquely mapped | 14 160 314 (87.75%) | 14 946 901 (88.18%) | 16 171 488 (87.67%) | 15 177 343 (88.27%) | 13 570 895 (88.47%) | 14 123 157 (88.50%) |
蛋白组测序后利用可信蛋白的表达量进行主成分分析(PCA分析),从不同维度展现样品间的关系。测序的两组样品(ES和MS)重复性较好,样品间差异大,可用于后期的数据分析(图2)。蛋白质组学数据已通过iProX存入ProteomeXchange Consortium(
图2
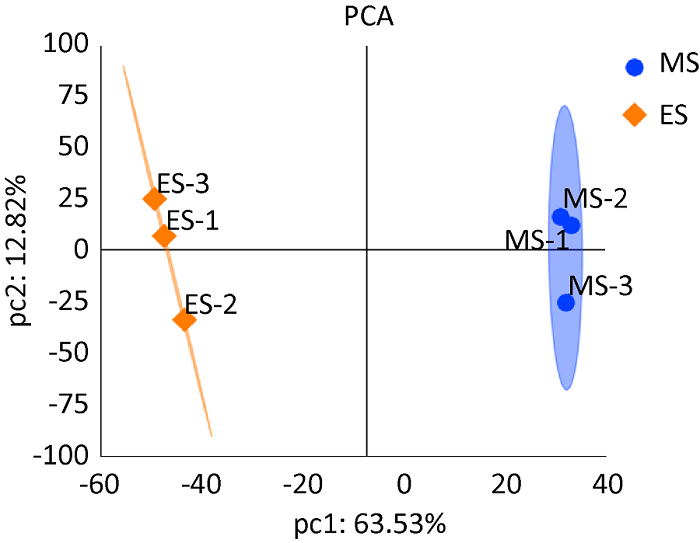
图2
可信蛋白表达量主成分分析
Fig. 2
Principal component analysis of credible protein expression.
2.2 差异基因和蛋白的筛选
转录组测序后,筛选得到差异表达基因721个,其中在成熟期菌柄时期上调表达基因为489个,下调表达基因232个。对差异表达基因构建火山图(volcano plot),能够清晰展示差异表达基因在伸长期菌柄与成熟期菌柄中的表达水平,以及差异的统计学显著性。图中的每一个点表示一个基因,横坐标表示基因在两样品中表达量差异倍数的对数值;纵坐标表示基因表达量变化的统计学显著性的负对数值。因此横坐标绝对值越大,说明表达量在两样品间的表达量倍数差异越大;纵坐标值越大,表明差异表达越显著,筛选得到的差异表达基因越可靠。从金针菇伸长期菌柄阶段到成熟期菌柄阶段,主要存在基因上调表达(图3A、4A)。其中上调表达中差异倍数对数值在|log2 Ratio|≥3的基因有51个,同水平下调表达基因仅有13个。
图3
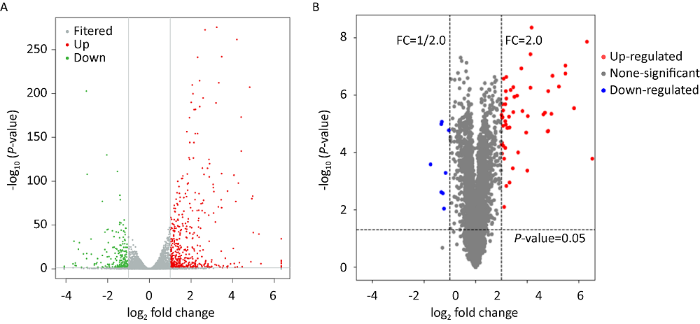
图3
差异表达基因和蛋白分布火山图
A:差异表达基因;B:差异表达蛋白
Fig. 3
Distribution volcano map of differentially expressed genes and protein.
A: Differentially expressed genes; B: Differentially expressed protein.
图4
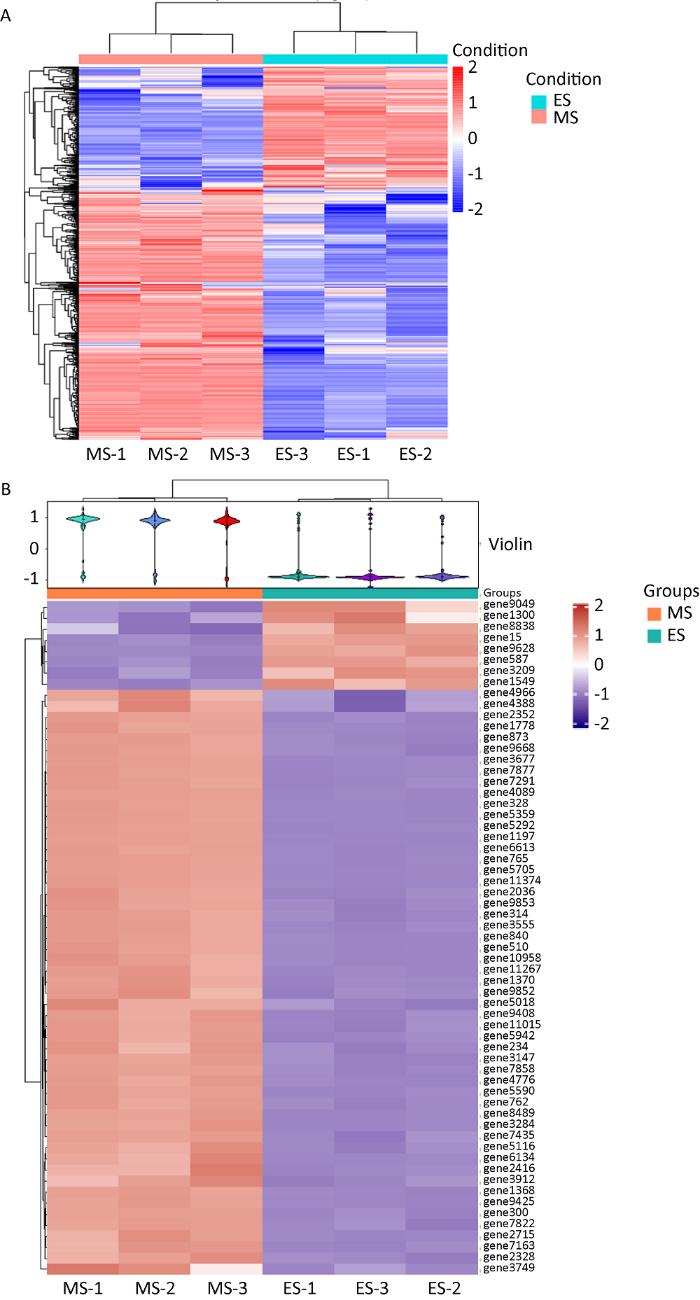
图4
差异表达基因和蛋白热敏图
A:差异表达基因;B:差异表达蛋白
Fig. 4
Thermosensitive maps of differentially expressed genes and proteins.
A: Differentially expressed genes; B: Differentially expressed proteins.
图5
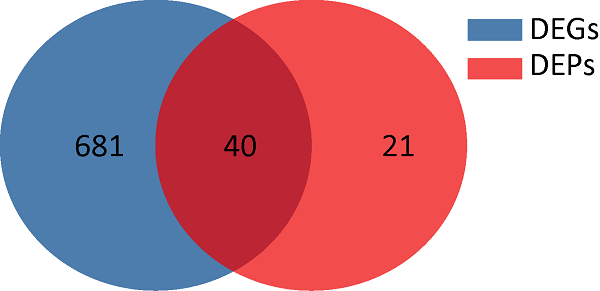
图5
差异表达基因和蛋白韦恩图
DEGs:差异表达基因;DEPs:差异表达蛋白
Fig. 5
Venn diagram of differentially expressed genes and proteins.
DEGs: Differentially expressed genes; DEPs: Differentially expressed protein.
2.3 差异表达基因和蛋白的GO功能聚类
为了进一步分析金针菇菌柄发育过程中差异表达基因所属功能,将全部差异基因和蛋白分别进行了GO功能聚类分析。GO分类单位有3个本体(ontology),分别描述基因的分子功能(molecular function)、细胞组分(cellular component)和参与的生物过程(biological process)。721个差异表达基因有290个被注释到42个GO二级单元条目中。其中值得注意的是差异基因数目达到总差异基因40%以上的条目有:催化活性(catalytic activity,72.41%)、细胞(cell,58.28%)、细胞组分(cell part,58.28%)、细胞过程(cellular process,52.41%)、绑定结合(binding,52.07%)、代谢过程(metabolic process,49.66%)、单生物过程(single-organism process,48.97%)(图6)。GO功能聚类结果暗示了这些条目中基因参与了金针菇菌柄的发育过程。
图6
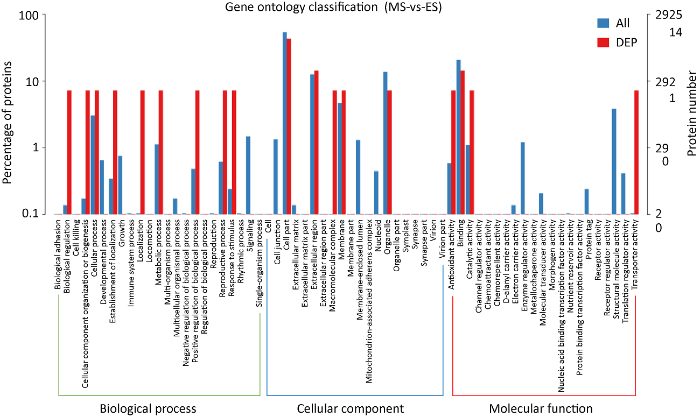
图6
差异表达基因GO功能聚类
Fig. 6
GO functional clustering of differentially expressed genes.
蛋白组数据分析表明,61个差异表达蛋白有14个被注释到18个GO 二级单元条目中。其中注释到差异表达蛋白较多的条目有:细胞组分(Cell part,6个)、绑定结合(Binding,2个)、胞外区(extracellular region,2个),其他条目均为1个差异表达蛋白(图7)。结合转录组数据,细胞组分和绑定结合条目同时被注释到较多的差异表达基因和蛋白。
图7
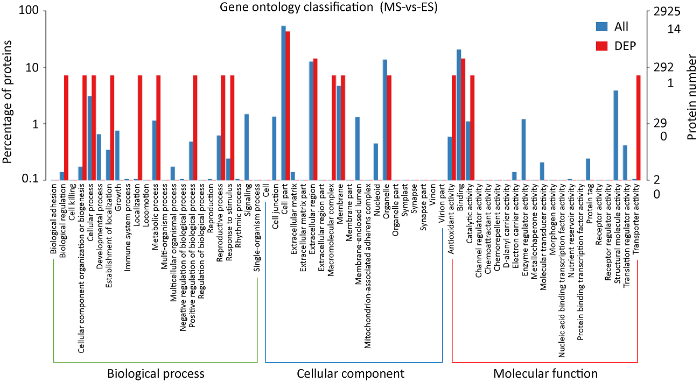
图7
差异表达蛋白GO功能聚类
Fig. 7
GO functional clustering of differentially expressed proteins.
2.4 差异表达基因和蛋白的KEGG pathway富集分析
KEGG是系统分析生命体各种新陈代谢和信息转导等pathway的信息数据库。转录组测序结果表明,成熟期与伸长期菌柄样品间共有96个差异表达基因被注释到20条通路内。其中在代谢通路中发现碳水化合物代谢通路(carbohydrate metabolism)所富集的基因最多,有19个基因(上调:17个;下调:2个)。其次是氨基酸代谢通路(amino acid metabolism)有11个基因(上调:9个;下调:2个)。从KEGG富集分析top20(筛选对应差异基因数目大于2的pathway条目,按照每个条目对应的-log10 P-value由大到小排序)气泡图可以看出:淀粉和蔗糖代谢(starch and sucrose metabolism,ko00500)、群体感应(quorum sensing,ko02024)、内质网内蛋白质加工(protein processing in endoplasmic reticulum,ko04141)、MAPK信号通路(MAPK signaling pathway-yeast,ko04011)、氨基酸生物合成(biosynthesis of amino acids,ko01230)通路富集基因较多(图8)。进一步分析发现,在差异表达基因富集最多的淀粉和蔗糖代谢途径中,海藻糖合成途径中的酶都被显著上调表达。
图8
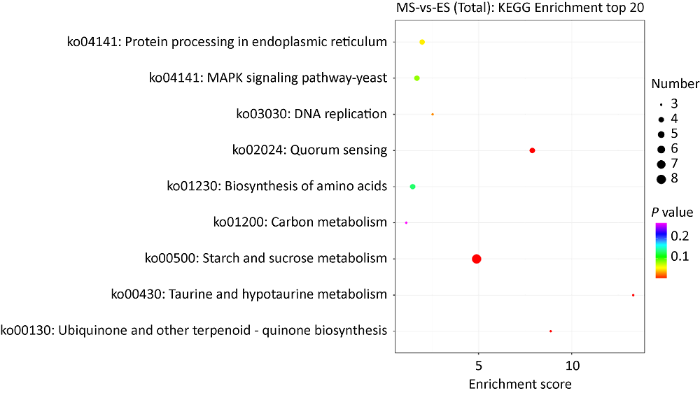
图8
差异表达基因KEGG富集分析
Fig. 8
KEGG enrichment analysis of differentially expressed genes.
蛋白组测序结果表明,成熟期与伸长期菌柄样品间共有11个差异表达蛋白被注释到11条通路上。从KEGG富集分析气泡图可以看出,富集分数较高的有:单环菌素生物合成(monobactam biosynthesis,ko00261)、链霉素生物合成(streptomycin biosynthesis,ko00521)、有机含硒化合物代谢(selenocompound metabolism,ko00450)等(图9)。值得注意的是内质网内蛋白质加工(protein processing in endoplasmic reticulum,ko04141)和MAPK信号通路(MAPK signaling pathway - yeast,ko04011)两个通路在转录组和蛋白组的KEGG富集分析中均为差异通路。
图9
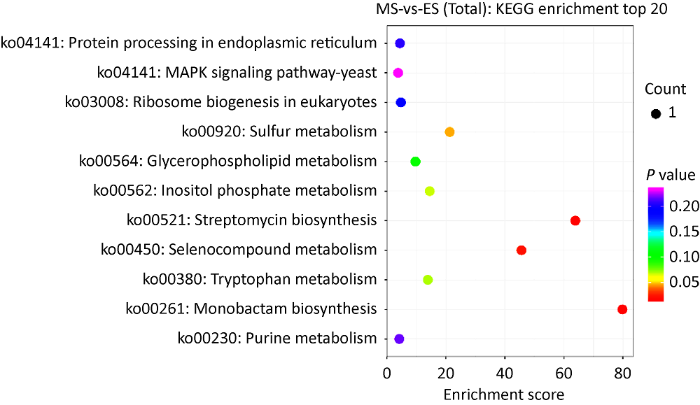
图9
差异表达蛋白KEGG富集分析
Fig. 9
KEGG enrichment analysis of differentially expressed proteins.
3 讨论
目前市场上有海鲜菇、蟹味菇、滑子菇、杏鲍菇、金针菇等多种食用菌,均以子实体菌柄作为重要的商品性状以及主要的可食用部分。因此,关于食用菌菌柄发育的机制研究具有很大的科学价值。本研究通过分析金针菇伸长期菌柄与成熟期菌柄间的转录组数据,筛选了上调以及下调表达的基因数量分别为489个和232个。其中上调表达中差异倍数对数值|log2 Ratio|≥3的基因有51个,同水平下调表达中为13个,因此在金针菇菌柄的发育过程中,主要存在基因的上调表达。此外,伸长期菌柄与成熟期菌柄的差异基因数目仅为721个,在金针菇同核体菌丝与异核体菌丝转录本中差异基因数目为3 504个,金针菇菌丝期与原基期差异基因数目为3 310个(刘芳等 2014;王威等 2015)。我们推测,可能因为菌柄从伸长期到成熟期发育过程中不存在较大的形态特征差异,所以筛选到的差异表达基因也比较少。主要在细胞组分、细胞过程、代谢过程等条目中富集了较多的差异表达基因或蛋白。
酶类作为一种重要的蛋白质,具有特异性及高效性,参与调控机体各项生化过程。吴光美等(2014)通过荧光定量技术发现金针菇漆酶家族基因Fv-lac4在金针菇菌柄部位的表达量显著高于菌盖。转录组测序技术也表明了金针菇漆酶家族基因lac1-lac11对于金针菇子实体发育具有重要作用(Wang et al. 2015)。姜思源等(2018)发现金针菇腺苷酸酶超家族基因Fv-Afe1在菌柄组织中高表达,并且伸长期菌柄的表达量显著高于成熟期菌柄。本研究发现在GO功能富集当中有72.41%的差异表达基因被聚类到分子功能下二级功能催化活性条目当中,这说明在菌柄由伸长期到成熟期的发育过程中,具有催化活性编码各类酶的基因起到重要作用。此外,一部分差异表达基因和蛋白被富集到Binding活性功能条目,该二级条目下的基因大多为转录因子。转录因子是一类具有“分子开关”作用的蛋白,通过与基因的上游调控序列进行特异性结合从而可以调节一系列基因的表达。陈仁良等(2016)发现金针菇凝集素基因Fv-mJRL1与其潜在的转录因子基因Fv-Mbp1之间存在极强的共表达规律。说明在金针菇菌柄的发育过程中,转录因子基因的差异表达会进一步调控下游基因表达。
KEGG可以提供经过整合的代谢途径注释,包括碳水化合物、核苷、氨基酸等有机物的代谢和降解途径(Hashimoto et al. 2006)。因此我们通过KEGG数据库对差异表达基因和蛋白进行富集分析,以期找出相关调控菌柄生长发育机制的通路。本研究发现,碳水化合物代谢通路和氨基酸代谢通路中富集了较多的差异表达基因,并且多为上调表达。这说明从菌柄的伸长期到成熟期过程中,能量代谢和氨基酸代谢都较为活跃。可能因为菌柄伸长时期是子实体快速发育的活跃时期,能量和氨基酸代谢活动都处于较高水平。值得注意的是,在差异表达基因富集最多的淀粉和蔗糖代谢途径中,海藻糖合成途径中的酶都被显著上调表达。有研究表明,在食用菌的栽培中,海藻糖的添加能够有效提高菌丝的生长和抗逆能力,甚至也能够提高食用菌的产量(Shirasaka et al. 2006;刘兵 2017;杨焕玲等 2019)。差异蛋白富集分数较高的有:单环菌素生物合成、链霉素生物合成和有机含硒化合物代谢。单环菌素和链霉素合成的上调,有可能是为了子实体在快速发育时期增强对有害微生物的抵抗能力。有机含硒化合物代谢在菌柄中活跃,能够为以后探索食用菌富硒新品种提供一定参考。和转录组KEGG富集相同,内质网蛋白质加工和MAPK信号通路在蛋白组的KEGG富集分析中也表现出差异。内质网是蛋白合成与成熟的重要场所,这表明在菌柄发育过程中,除了能量代谢活跃,蛋白合成过程也较为活跃。作为子实体的重要部位,菌柄需要感知重力、二氧化碳浓度、光照等环境因素,从而调整子实体的发育形态。MAPK信号通路是真核生物信号传递网络中的重要途径之一,在基因表达调控和细胞质功能活动中发挥关键作用。
本研究基于转录组与蛋白组测序技术,分析了成熟期和伸长期菌柄的差异表达基因和蛋白。这为我们下一步研究金针菇及其他食用菌的菌柄发育分子调控机制提供思路与参考。下一步将结合基因功能研究方法对相关基因进行功能检验,以期解析食用菌的菌柄发育过程。
致谢
感谢福建省食用菌种质资源保藏与管理中心提供金针菇菌株F19;感谢山东农业大学菌物实训实践基地提供试验条件。
参考文献
HTSeq-a Python framework to work with high-throughput sequencing data
DOI:10.1093/bioinformatics/btu638
PMID:25260700
[本文引用: 1]

A large choice of tools exists for many standard tasks in the analysis of high-throughput sequencing (HTS) data. However, once a project deviates from standard workflows, custom scripts are needed.We present HTSeq, a Python library to facilitate the rapid development of such scripts. HTSeq offers parsers for many common data formats in HTS projects, as well as classes to represent data, such as genomic coordinates, sequences, sequencing reads, alignments, gene model information and variant calls, and provides data structures that allow for querying via genomic coordinates. We also present htseq-count, a tool developed with HTSeq that preprocesses RNA-Seq data for differential expression analysis by counting the overlap of reads with genes.HTSeq is released as an open-source software under the GNU General Public Licence and available from http://www-huber.embl.de/HTSeq or from the Python Package Index at https://pypi.python.org/pypi/HTSeq.© The Author 2014. Published by Oxford University Press.
The significance of digital gene expression profiles
Genes differentially expressed in different tissues, during development, or during specific pathologies are of foremost interest to both basic and pharmaceutical research. "Transcript profiles" or "digital Northerns" are generated routinely by partially sequencing thousands of randomly selected clones from relevant cDNA libraries. Differentially expressed genes can then be detected from variations in the counts of their cognate sequence tags. Here we present the first systematic study on the influence of random fluctuations and sampling size on the reliability of this kind of data. We establish a rigorous significance test and demonstrate its use on publicly available transcript profiles. The theory links the threshold of selection of putatively regulated genes (e.g., the number of pharmaceutical leads) to the fraction of false positive clones one is willing to risk. Our results delineate more precisely and extend the limits within which digital Northern data can be used.
Bioinformatic analysis and co-expression study of jacalin-related lectin gene Fv-mJRL1 in Flammulina velutipes
Non-coding RNAs: meet thy masters
Notes on the nomenclature of five important edible fungi in China
Species diversity of Flammulina in China: new varieties and a new record
Growth and rotation of Flammulina velutipes fruit bodies and the dependence of stipe elongation on the cap
DOI:10.2307/3757353 URL [本文引用: 1]
Promotion of stipe elongation in isolated Flammulina velutipes fruit bodies by carbohydrates, natural extracts, and amino acids
DOI:10.1139/b72-097 URL [本文引用: 1]
Effects of CO2 concentration on the growth and development of Flammulina velutipes
KEGG as a glycome informatics resource
DOI:10.1093/glycob/cwj010 URL [本文引用: 1]
Identification and expression patterns of fvexpl1, an expansin-like gene, suggests an auxiliary role in the stipe morphogenesis of Flammulina velutipes
DOI:10.4014/jmb.1712.12046 URL [本文引用: 1]
Preliminary study on the function of Fv-Afe1 gene in Flammulina velutipes
KEGG for linking genomes to life and the environment
Further characterization of hydrophobin genes in genome of Flammulina velutipes
DOI:10.1016/j.myc.2016.04.004 URL [本文引用: 1]
Distribution of cellular carbohydrates during development of the mycelium and fruitbodies of Flammulina velutipes
Flammulina velutipes (Curt. ex Fr.) Sing. was grown on potato-glucose solution freed of most starch. Glucose uptake and dry weight changes in the colony indicated that the large fruitbodies derived their substrates partly from glucose remaining in the medium and partly from cellular constituents stored in the mycelium and small fruitbodies. Changes in the amounts of low molecular weight carbohydrates, glycogen, and four cell wall polysaccharide fractions were followed in the mycelium and fruitbodies. Trehalose, arabitol, and smaller amounts of mannitol were the main stored low molecular weight carbohydrates. A large net loss of these compounds occurred in the mycelium and small fruitbodies after their growth ceased. The carbohydrates accumulated in the large fruitbodies, but were also partly metabolized in the colony. Reducing sugars were minor components, and included about 30 to 50% glucose and a small undetermined quantity of fructose. Glycogen was the main storage carbohydrate in the mycelium, and was also stored in the small fruitbodies. It was broken down in both structures during growth of the large fruitbodies which accumulated only small amounts. During the same period, almost 45% of the maximum amount of cell wall polysaccharides were degraded in the small fruitbodies, but not in the mycelium.By feeding (14)C-glucose in replacement medium, incorporation of radioactivity into carbohydrates was followed in the colony during fruit-body development. Total incorporation was highest in trehalose, next highest in glycogen, and the rest was found in polyols and cell wall polysaccharides except for a few per cent which remained in endogenous glucose. In the large fruitbodies, specific radioactivity in glucose was much lower than in trehalose and mannitol. The labeling patterns in the mycelium and large fruitbodies suggested that trehalose, mannitol, and possibly arabitol were translocated into the stipes and pilei.
Effect of GABA and trehalose on the activity of exoenzyme induced by heat stress in mycelia of Lentinus edodes
Comparison of gene expression patterns in the mycelium and primordia of Flammulina velutipes
A comparative proteome approach reveals metabolic changes associated with Flammulina velutipes mycelia in response to cold and light stress
DOI:10.1021/acs.jafc.8b00383 URL [本文引用: 1]
iTRAQ-based quantitative proteome revealed metabolic changes of Flammulina velutipes mycelia in response to cold stress
DOI:10.1016/j.jprot.2017.01.009 URL
The influence of factors of the aeration complex and light upon fruit-body form in pure cultures of an agaric and a polypore
DOI:10.1093/oxfordjournals.aob.a083543 URL [本文引用: 1]
Identification of novel transcripts in annotated genomes using RNA-Seq
DOI:10.1093/bioinformatics/btr355 URL [本文引用: 1]
Improving RNA-Seq expression estimates by correcting for fragment bias
Influence of light on the morphological changes that take place during the development of the Flammulina velutipes fruit body
DOI:10.1007/S10267-004-0195-7 URL [本文引用: 1]
Changes in the CO2 and amount of mycelium growth of the liquid spawn on Flammulina velutipes
Effect of trehalose on the heat tolerance of Lentinula edodes mycelia
Transcriptomics and functional genomics of plant defence induction by phloem-feeding insects
The relationship between phloem-feeding insects (PFIs) and plants offers an intriguing example of a highly specialized biotic interaction. These insects have evolved to survive on a nutritionally imbalanced diet of phloem sap, and to minimize wound responses in their host plants. As a consequence, plant perception of and responses to PFIs differ from plant interactions with other insect-feeding guilds. Transcriptome-wide analyses of gene expression are currently being applied to characterize plant responses to PFIs in crop plants with race-specific innate resistance, as well as in compatible interactions with susceptible hosts. Recent studies indicate that PFIs induce transcriptional reprogramming in their host plants, and that plant responses to PFIs appear to be quantitatively and qualitatively different from responses to other insects or pathogens. Transcript profiling studies also suggest that PFIs induce cell wall modifications, reduce photosynthetic activity, manipulate source-sink relations, and modify secondary metabolism in their hosts, and many of these responses appear to occur within the phloem tissue. Plant responses to these insects appear to be regulated in part by the salicylate, jasmonate, and ethylene signalling pathways. As additional transcript profiling data become available, forward and reverse genetic approaches will be necessary to determine which changes in gene expression influence resistance or susceptibility to PFIs.
Characterization of the yeast transcriptome
We have analyzed the set of genes expressed from the yeast genome, herein called the transcriptome, using serial analysis of gene expression. Analysis of 60,633 transcripts revealed 4,665 genes, with expression levels ranging from 0.3 to over 200 transcripts per cell. Of these genes, 1981 had known functions, while 2684 were previously uncharacterized. The integration of positional information with gene expression data allowed for the generation of chromosomal expression maps identifying physical regions of transcriptional activity and identified genes that had not been predicted by sequence information alone. These studies provide insight into global patterns of gene expression in yeast and demonstrate the feasibility of genome-wide expression studies in eukaryotes.
Phylogeny and species delimitation of Flammulina: taxonomic status of winter mushroom in East Asia and a new European species identified using an integrated approach
DOI:10.1007/s11557-018-1409-2 URL [本文引用: 1]
Comparison of gene expression patterns between the monokaryotic and dikaryotic mycelia of Flammulina velutipes
The multigene family of fungal laccases and their expression in the white rot basidiomycete Flammulina velutipes
DOI:10.1016/j.gene.2015.03.020
PMID:25776201
Fungal laccases play important roles in matrix degradation. Eleven laccase genes, including three novel ones (designated lac1, lac2 and lac4) were identified after sequencing the entire genome of the edible, white-rot fungus Flammulina velutipes. Analysis using bioinformatics revealed that all of the laccases, except lac3, possess a signal peptide. These laccase proteins consist of 502-670 amino acids and have predicted molecular weights ranging from 55kDa to 74kDa. These proteins each contain four copper-binding sites, except for Lac10. Transcriptomes were sequenced at different developmental stages and in different fruiting body tissues to analyze if there was differential expression of laccase genes. The novel laccase gene lac4 exhibited the highest expression levels among all of the observed laccases at every developmental stage and in all fruiting body tissues examined. We conclude that laccases in F. velutipes play a role not only in lignin degradation, but also in fruiting body formation and development.Copyright © 2015 Elsevier B.V. All rights reserved.
Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize
DOI:10.1105/tpc.109.065714 URL [本文引用: 1]
Analysis on structure and differential expression of a laccase gene Fv-lac4 that possible involves in development of fruit body of Flammulina velutipes
Effect of adding trehalose to culture medium on the growth of Stropharia rugosoannulata and Hypsizigus marmoreus
A specific protein, flammutoxin expressed in the Flammulina velutipes stipe
Effects of extraction methods on the antioxidant activities of polysaccharides obtained from Flammulina velutipes
DOI:10.1016/j.carbpol.2013.07.052 URL [本文引用: 1]
金针菇Fv-Afe1基因的功能初探
DOI:10.13560/j.cnki.biotech.bull.1985.2018-0019
[本文引用: 2]
从金针菇菌株L11基因组数据筛选出一个腺苷酸合成酶超家族基因Fv-Afe1。结合转录组数据分析基因结构,利用生物信息学软件对基因及其氨基酸序列生理生化指标进行预测,与NCBI已公布的其他物种的氨基酸序列进行系统发育分析,运用qRT-PCR技术和转录组表达谱数据对Fv-Afe1在金针菇各发育时期进行表达分析。结果显示,Fv-Afe1全长为967 bp,具有4个外显子和3个内含子,该基因氨基酸三级结构具有两个结构域,属于胞内酶,无跨膜结构,为真菌腺苷酸合成酶超家族成员;该基因在伸长期菌柄高表达,成熟期菌柄次之。
与金针菇子实体发育有关的漆酶编码基因Fv-lac4的结构及表达差异分析



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}