PDF(755 KB)
PDF(755 KB)
 PDF(755 KB)
PDF(755 KB)
PDF(755 KB)
PDF(755 KB)
沉水玛利亚霉新种以及链状玛利亚霉的新生境记录
 ({{custom_author.role_cn}}), {{javascript:window.custom_author_cn_index++;}}
({{custom_author.role_cn}}), {{javascript:window.custom_author_cn_index++;}}Mariannaea submersa sp. nov., with a new habitat and geographic record of Mariannaea catenulata
({{custom_author.role_en}}), {{javascript:window.custom_author_en_index++;}}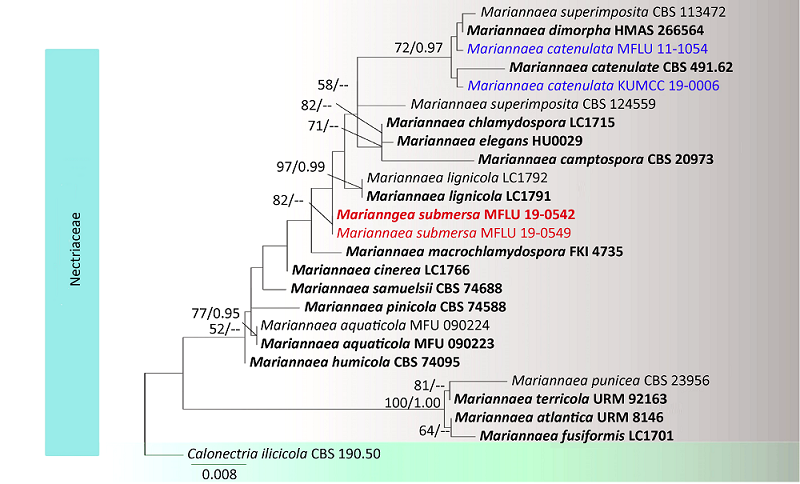
| {{custom_ref.label}} |
{{custom_citation.content}}
{{custom_citation.annotation}}
|


/
| 〈 |
|
〉 |