PDF(1014 KB)
PDF(1014 KB)

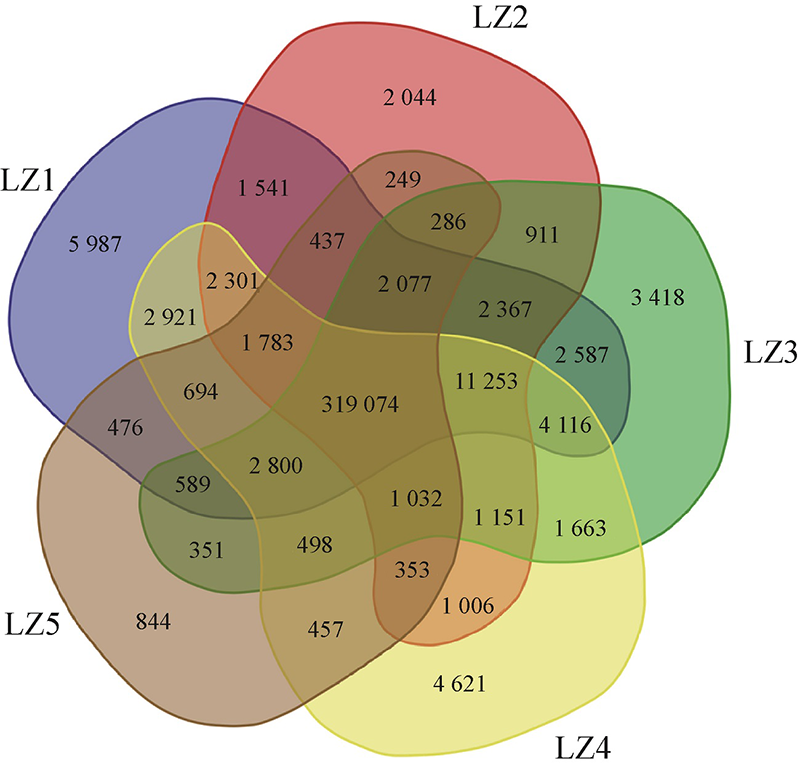
Establishment of molecular markers in the whole genome of Ganoderma lingzhi elite varieties
LIU Yiting, JIANG Xiaohan, YANG Chunyan, CHEN Jianhui, WANG Chezhao, LÜ Xiaomeng, YANG Zhikang, DENG Youjin, WU Xiaoping
Mycosystema ›› 2025, Vol. 44 ›› Issue (3) : 240141.
PDF(1014 KB)
PDF(1014 KB)
Establishment of molecular markers in the whole genome of Ganoderma lingzhi elite varieties
 ({{custom_author.role_en}})
,
{{javascript:window.custom_author_en_index++;}}
({{custom_author.role_en}})
,
{{javascript:window.custom_author_en_index++;}}
| {{custom_ref.label}} |
{{custom_citation.content}}
{{custom_citation.annotation}}
|


/
| 〈 |
|
〉 |