PDF(1206 KB)
PDF(1206 KB)

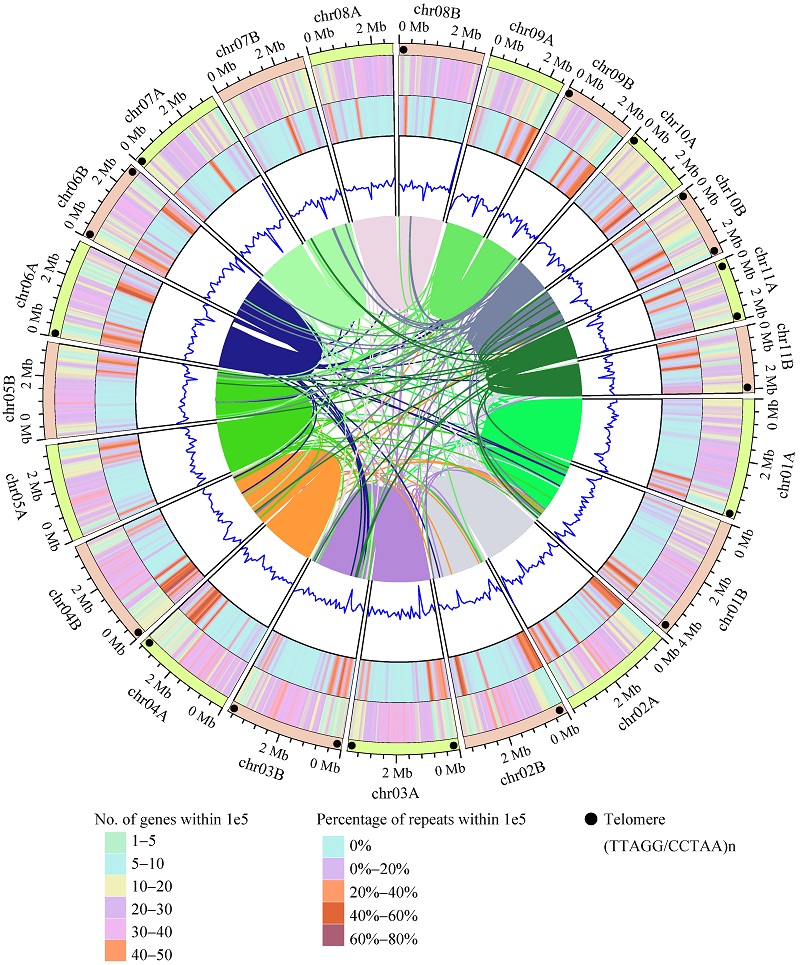
Typification and genome analyses of early described fungal species: a case study of Lysurus mokusin, the first new fungal species described in China
LIANG Junmin, WANG Ke, DU Zhuo, ZHAO Mingjun, CAI Lei, DAI Yucheng
Mycosystema ›› 2025, Vol. 44 ›› Issue (4) : 240296.
PDF(1206 KB)
PDF(1206 KB)
Typification and genome analyses of early described fungal species: a case study of Lysurus mokusin, the first new fungal species described in China
 ({{custom_author.role_en}})
,
{{javascript:window.custom_author_en_index++;}}
({{custom_author.role_en}})
,
{{javascript:window.custom_author_en_index++;}}
| {{custom_ref.label}} |
{{custom_citation.content}}
{{custom_citation.annotation}}
|


/
| 〈 |
|
〉 |